What To Know About FDA Medical Device Classes?
Written by
Arterex Medical
Published on
October 26, 2024
Read time
12 minute read
The FDA’s classification system for medical devices plays a crucial role in ensuring patient safety and product effectiveness. This system categorizes devices into three classes—Class I, II, and III—based on their risk level and the regulatory controls needed to ensure their safety.
Class I devices pose the lowest risk and are subject to the least stringent controls, while Class III devices present the highest risk and undergo the most rigorous scrutiny. This classification determines the regulatory pathway a device must follow, from general controls for Class I to premarket approval for most Class III devices.

Understanding these classifications is essential for manufacturers, healthcare providers, and patients alike. The system covers a wide range of medical devices, from simple bandages to complex implantable pacemakers, and balances the need for innovation with the paramount concern for public health and safety.
What are FDA Medical Device Classes?
The U.S. Food and Drug Administration (FDA) regulates all medical devices marketed in the United States. Through its Center for Devices and Radiological Health, the FDA assigns every single medical device a ‘class’ based on its risk profile – that is, the potential threat posed to the safety and health of a patient were something to go wrong with it.
The FDA categorizes medical devices into three distinct classes:
- Class I Devices:
- Lowest risk devices
- Examples: bandages, tongue depressors, and handheld surgical instruments
- Subject to general controls, such as good manufacturing practices and proper labeling
- Most Class I devices are exempt from premarket notification (510(k)) requirements
- Class II Devices:
- Moderate risk devices
- Examples: powered wheelchairs, infusion pumps, and surgical drapes
- Subject to general controls and special controls, which may include performance standards, post-market surveillance, and specific labeling requirements
- Most Class II devices require a 510(k) premarket notification
- Class III Devices:
- Highest risk devices
- Examples: implantable pacemakers, breast implants, and deep brain stimulators
- Subject to general controls and premarket approval (PMA)
- Require clinical data to demonstrate safety and effectiveness before market approval
The classification of a medical device determines the regulatory pathway it must follow before being marketed in the United States. As the class level increases, so does the level of FDA scrutiny and the amount of evidence required to demonstrate the device’s safety and effectiveness.
It’s important to note that there’s an enormous difference in the optimal paths to market, depending on how a device is grouped. Class I devices are subject to far fewer regulatory requirements than Class II or III devices. This classification system ensures that the level of regulatory control is appropriate to the level of potential risk associated with each device.
The lines between these classes are drawn based on the device’s invasiveness, its potential impact on patient health, and the overall risk it poses. This system allows the FDA to allocate its resources effectively, focusing more intensively on higher-risk devices while maintaining appropriate oversight of lower-risk products.
What Are Examples of Medical Device Classes?
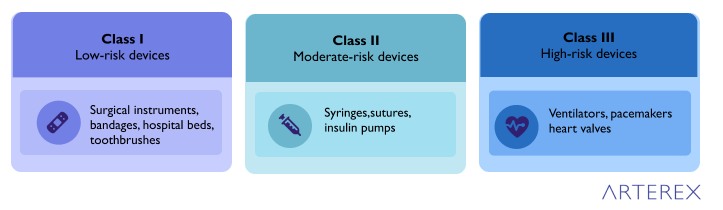
To better understand the FDA’s classification system, it’s helpful to examine examples of medical devices in each risk category. This overview brings the class hierarchy into focus, illustrating how the level of risk and potential impact on patient health correlates with the device classification.
Class I Devices (Low Risk):
- Bandages and adhesive strips
- Toothbrushes and dental floss
- Hand-held surgical instruments (e.g., scalpels, forceps)
- Tongue depressors
- Elastic bandages
- Examination gloves
- Manual stethoscopes
Class II Devices (Moderate Risk):
- Syringes
- Insulin pumps
- Blood pressure cuffs
- Powered wheelchairs
- Pregnancy test kits
- Surgical drapes
- Infusion pumps
- Contact lenses
Class III Devices (High Risk):
- Ventilators
- Implantable pacemakers
- Breast implants
- Deep brain stimulators
- Artificial heart valves
- Cochlear implants
- Implantable defibrillators
This medical device list demonstrates how the FDA categorizes devices based on their potential risk and impact on patient health. Low-risk devices with minimal patient impact, such as bandages and toothbrushes, fall into Class I. These devices generally pose little risk even if they malfunction.
Class II encompasses medium-risk devices like syringes and insulin pumps. While these devices are more complex and can have a more significant impact on patient health, they are generally well-understood in terms of technology and use.
High-risk devices in Class III, such as ventilators and implantable pacemakers, are often life-sustaining or life-supporting. They may be highly invasive or play a critical role in maintaining a patient’s health, which is why they undergo the most rigorous FDA scrutiny before approval.
Understanding these examples helps illustrate why different devices require varying levels of regulatory oversight to ensure patient safety and product effectiveness.
What Are The Different Types of Medical Devices?
Medical devices encompass a wide range of products used in healthcare settings. While the FDA classifies devices based on risk, it’s also helpful to categorize them by their function and application. Here are the main types of medical devices:
Diagnostic Devices:
- Used to detect, diagnose, or monitor medical conditions
- Examples: X-ray machines, MRI scanners, blood glucose meters, thermometers
Therapeutic Devices:
- Used to treat medical conditions or provide therapy
- Examples: Infusion pumps, TENS units, dialysis machines, radiation therapy equipment
Life Support Devices:
- Critical for maintaining vital bodily functions
- Examples: Ventilators, heart-lung machines, defibrillators
Implantable Devices:
- Placed inside the body to replace or support organ function
- Examples: Pacemakers, artificial joints, coronary stents, cochlear implants
Surgical Devices:
- Used during surgical procedures
- Examples: Scalpels, surgical robots, laparoscopic instruments, surgical lasers
Prosthetic and Orthotic Devices:
- Replace or support body parts
- Examples: Artificial limbs, orthopedic braces, hearing aids
Monitoring Devices:
- Track and record patient vital signs or other health metrics
- Examples: ECG monitors, pulse oximeters, wearable fitness trackers
Dental Devices:
- Used in dental procedures and oral care
- Examples: Dental implants, braces, dental x-ray equipment
Ophthalmic Devices:
- Related to eye care and vision correction
- Examples: Contact lenses, intraocular lenses, LASIK surgical equipment
In Vitro Diagnostic Devices:
- Used to perform tests on samples derived from the human body
- Examples: HIV test kits, pregnancy tests, genetic tests
These categories often overlap, and many devices may fall into multiple types. For instance, an implantable cardioverter-defibrillator (ICD) is both an implantable device and a life support device. The diversity of medical devices reflects the complexity of modern healthcare and the ongoing advancements in medical technology.
What is an FDA Class 1 Medical Device?
FDA Class I medical devices are defined as devices that are “not intended for use in supporting or sustaining life or of substantial importance in preventing impairment to human health, and they may not present a potential unreasonable risk of illness or injury.” These devices represent the lowest risk category in the FDA’s classification system and constitute approximately 47% of all FDA-regulated medical devices on the market.
Class I devices are characterized by their minimal contact with patients and low impact on a patient’s overall health. Generally, these devices do not come into contact with a patient’s internal organs, central nervous system, or cardiovascular system. Due to their low-risk nature, Class I devices are subject to the least stringent regulatory requirements among the three device classes.
The regulatory pathway for Class I devices is relatively straightforward. Most Class I devices are exempt from the FDA requirements for Premarket Notification (510(k)) and Premarket Approval (PMA). However, they are not exempt from FDA General Controls, which apply to all medical device classes. These General Controls address issues such as adulteration, misbranding, device registration, record-keeping, and good manufacturing practices.
While the regulatory burden is lower for Class I devices, manufacturers are still required to implement a quality management system and adhere to standards that ensure product quality. This approach allows the FDA to maintain appropriate oversight while allocating more resources to higher-risk devices.
Class 1 Medical Device Examples
- Electric toothbrush
- Tongue depressor
- Oxygen mask
- Reusable surgical scalpel
- Bandages and adhesive strips
- Hospital beds
- Non-electric wheelchair
- Elastic bandages
- Examination gloves
- Manual stethoscopes
These examples illustrate the low-risk nature of Class I devices. They are typically simple in design, have a well-established safety profile, and pose minimal risk to patients even if they malfunction.
What is an FDA Class 2 Medical Device?
FDA Class II medical devices are more complex than Class I devices and present a higher category of risk. The FDA defines Class II devices as those “for which general controls are insufficient to provide reasonable assurance of the safety and effectiveness of the device.” These devices are more likely to come into sustained contact with a patient, including contact with the cardiovascular system or internal organs. Class II also includes many diagnostic tools.
Class II devices require a higher level of regulatory control to ensure their safety and effectiveness. In addition to the General Controls that apply to all device classes, Class II devices are subject to Special Controls. These may include:
- Device performance standards
- Post-market surveillance
- Patient registries
- Special labeling requirements
- Premarket data requirements
- Specific guidelines
The regulatory pathway for most Class II devices involves the premarket notification 510(k) process. This process requires manufacturers to demonstrate that their device is substantially equivalent to a legally marketed predicate device. Substantial equivalence doesn’t mean the devices need to be identical, but they must have significant similarities in use, design, materials, labeling, standards, and other characteristics.
It’s worth noting that in early 2018, the FDA released an exemption list that exempts over 800 device types from the 510(k) process. Manufacturers of generic Class II medical devices should check if their device is exempt from 510(k) filing.
Class 2 Medical Device Examples
- Catheters
- Blood pressure cuffs
- Pregnancy test kits
- Syringes
- Blood transfusion kits
- Contact lenses
- Surgical gloves
- Absorbable sutures
- Powered wheelchairs
- Infusion pumps
These examples demonstrate the increased complexity and potential risk associated with Class II devices compared to Class I. While they may have more direct interaction with patients or play a more significant role in diagnosis or treatment, they are generally well-understood in terms of technology and use.
What is an FDA Class 3 Medical Device?

FDA Class III medical devices represent the highest risk category. The FDA defines Class III devices as products that “usually sustain or support life, are implanted, or present a potential unreasonable risk of illness or injury.” Only about 10% of FDA-regulated devices fall into this category, which typically includes permanent implants, smart medical devices, and life support systems.
Class III devices are subject to the most stringent regulatory controls. In addition to General Controls, most Class III devices must go through the Premarket Approval (PMA) process. The FDA states that “general and special controls alone are insufficient to assure the safety and effectiveness of Class III devices.”
The PMA is the most intensive type of device marketing application required by the FDA. It involves a rigorous study of the Class III medical device to prove safety and effectiveness through the development of a data-driven benefit/risk profile. This process generally involves clinical trials and requires significant time and resources for sufficient data collection.
In rare cases, some Class III devices may qualify for the 510(k) route if a suitable predicate marketed before the Medical Device Amendments of 1976 can be found. However, this pathway is becoming increasingly unlikely and rare, as the FDA discourages the use of predicates older than a decade.
During the PMA process, the FDA will also perform a substantive review of the manufacturer’s quality system. This comprehensive evaluation ensures that high-risk devices meet the strictest standards for safety and effectiveness before they reach the market.
Class 3 Medical Device Examples
- Breast implants
- Pacemakers
- Implantable cardioverter-defibrillators (ICDs)
- High-frequency ventilators
- Cochlear implants
- Fetal blood sampling monitors
- Implanted prosthetics
- Artificial heart valves
- Deep brain stimulators
- Implantable infusion pumps
These examples highlight the critical nature of Class III devices. They are often implanted in the body, sustain or support life, or have the potential to significantly impact a patient’s health and well-being. The high level of risk associated with these devices justifies the extensive regulatory scrutiny they undergo before and after market approval.
What Is the Difference Between Class 1, Class 2, and Class 3 Medical Devices?
The U.S. Food and Drug Administration (FDA) classifies medical devices into three categories based on their risk level and the regulatory controls necessary to ensure their safety and effectiveness. Understanding the differences between these classes is crucial for manufacturers, healthcare providers, and patients. Let’s explore the key distinctions:
Risk Level:
Class 1: Lowest risk
Class 2: Moderate risk
Class 3: Highest risk
The risk level is determined by factors such as the device’s intended use, invasiveness, and potential impact on patient health.
Regulatory Controls:
Class 1:
Subject to General Controls
Most exempt from Premarket Notification (510(k))
Typically do not require clinical data for approval
Class 2:
Subject to General Controls and Special Controls
Most require 510(k) submission
May require clinical data, depending on the device
Class 3:
Subject to General Controls and Premarket Approval (PMA)
Almost always require extensive clinical data
Examples:
Class 1: Bandages, tongue depressors, manual stethoscopes
Class 2: Powered wheelchairs, infusion pumps, surgical drapes
Class 3: Implantable pacemakers, breast implants, cochlear implants
Complexity and Innovation:
Class 1: Generally simple in design with well-established safety profiles
Class 2: More complex, often involving new technologies or modifications of existing devices
Class 3: Often cutting-edge technology or implantable devices
Patient Contact:
Class 1: Minimal patient contact or external use only
Class 2: May have sustained contact with patients, including some internal contact
Class 3: Often implanted or used to sustain life
Regulatory Pathway:
Class 1: Most exempt from premarket submission
Class 2: Most require 510(k) clearance, demonstrating substantial equivalence to a predicate device
Class 3: Typically require PMA, involving extensive safety and efficacy data
Manufacturing Requirements:
Class 1: Must adhere to Good Manufacturing Practices (GMP)
Class 2: Must follow GMP and may have additional quality system requirements
Class 3: Subject to the most stringent quality system regulations and manufacturing controls
Post-Market Surveillance:
Class 1: Minimal post-market surveillance required
Class 2: May require post-market surveillance, depending on the device
Class 3: Often subject to rigorous post-market surveillance and long-term studies
Time to Market:
Class 1: Quickest path to market
Class 2: Moderate time to market, depending on 510(k) review
Class 3: Longest time to market due to extensive PMA process
Regulatory Burden:
Class 1: Least burdensome regulatory requirements
Class 2: Moderate regulatory burden
Class 3: Most burdensome regulatory requirements
FDA Scrutiny:
Class 1: Least FDA scrutiny
Class 2: Moderate FDA scrutiny
Class 3: Highest level of FDA scrutiny throughout the device lifecycle
Potential for Harm:
Class 1: Minimal potential for harm if the device fails
Class 2: Moderate potential for harm if the device fails
Class 3: Significant potential for harm if the device fails, often life-threatening
Understanding these differences is crucial for navigating the regulatory landscape, ensuring compliance, and bringing medical devices to market efficiently and safely. The classification system allows the FDA to allocate its resources effectively, focusing more intensively on higher-risk devices while maintaining appropriate oversight of lower-risk products. This tiered approach balances the need for innovation in medical technology with the paramount concern for patient safety.
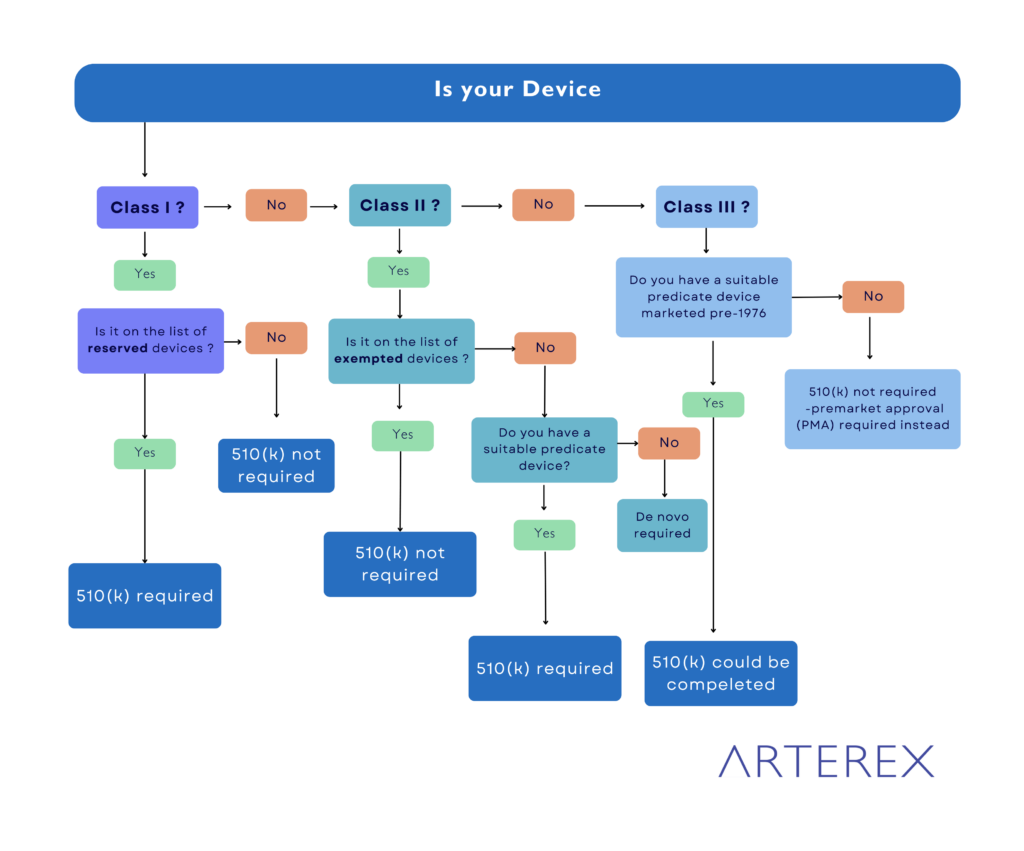
How To Classify a Medical Device?
Determining the correct classification for your medical device is a crucial first step in the FDA regulatory process. Here’s a step-by-step guide to help you navigate the classification system:
- Consult FDA Classification Regulations: Begin by examining the FDA’s list of 16 medical device categories, organized by medical specialization. These categories cover a wide range of devices from various fields of medicine.
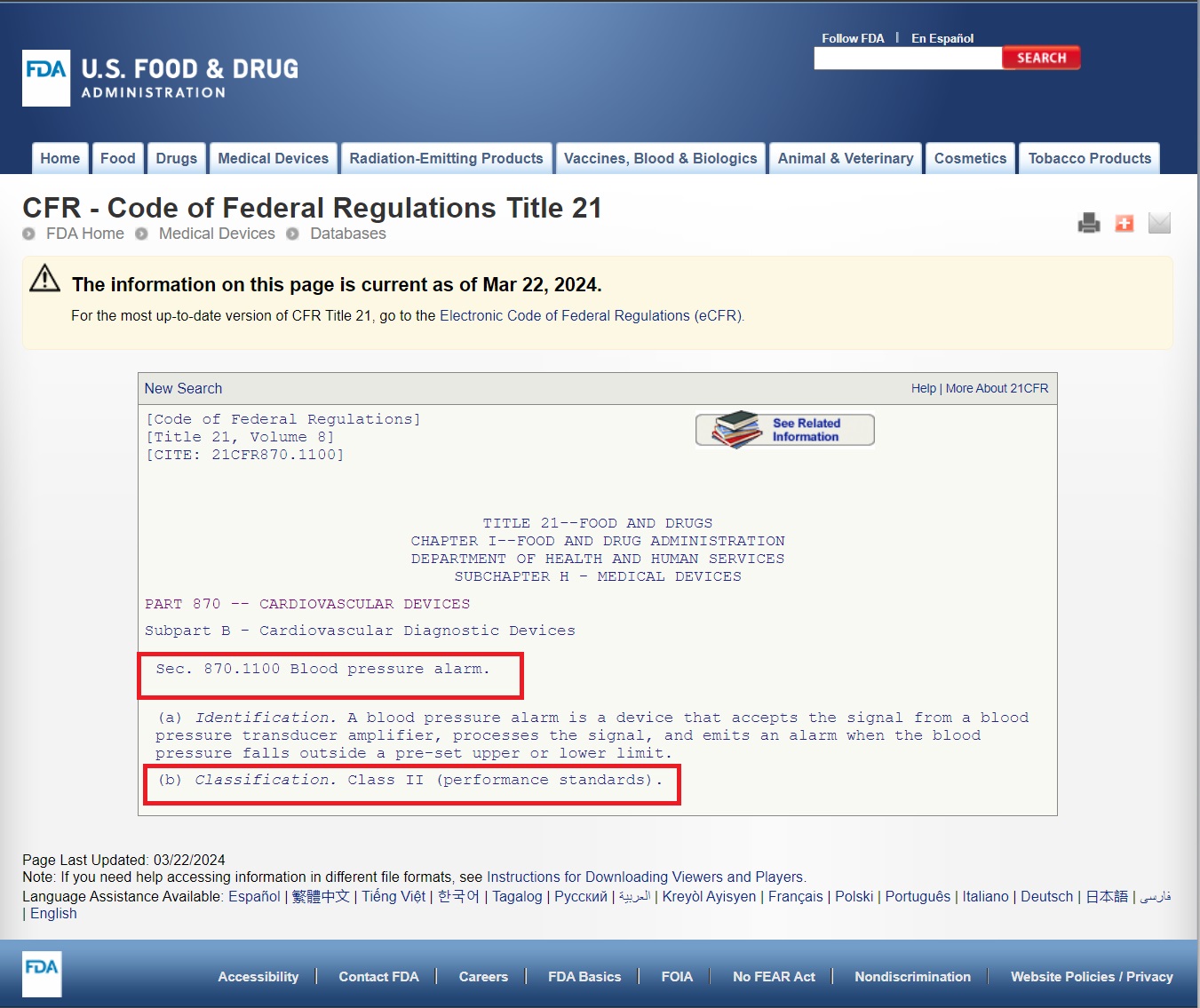
- Identify the Relevant Category: Determine which category best fits your device based on its intended use and medical specialty. For instance, a blood pressure alarm would fall under Category 870: Cardiovascular Devices.
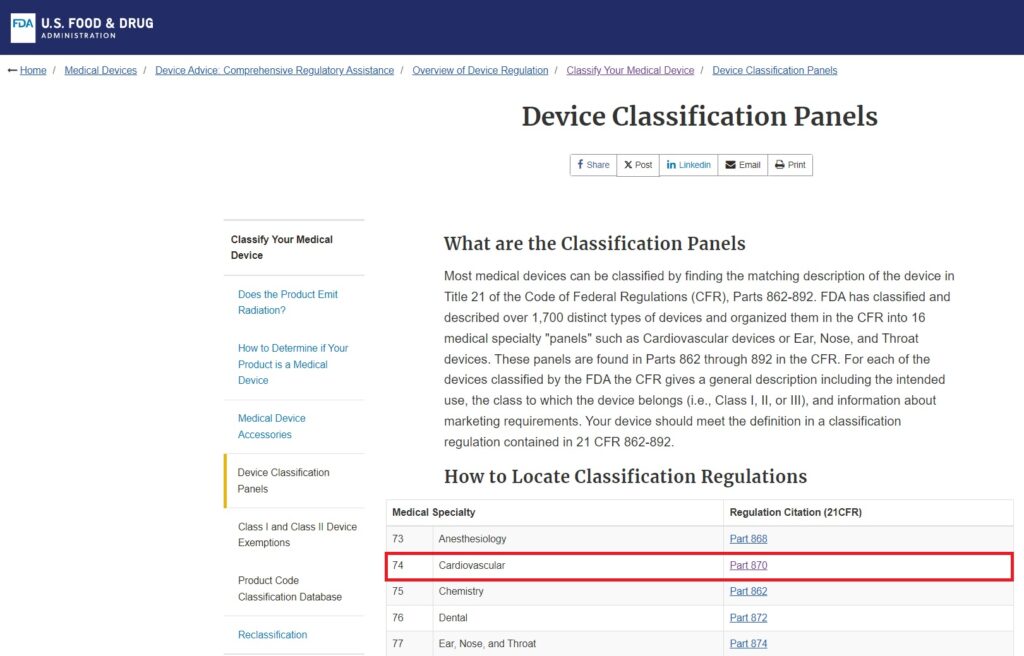
3 . Search for Similar Devices: Within the chosen category, carefully review the list of devices to find one that closely matches your product’s function and characteristics.
4. Locate the Device Code: Once you’ve found a similar device, note its associated device code. This code is crucial for further classification information.
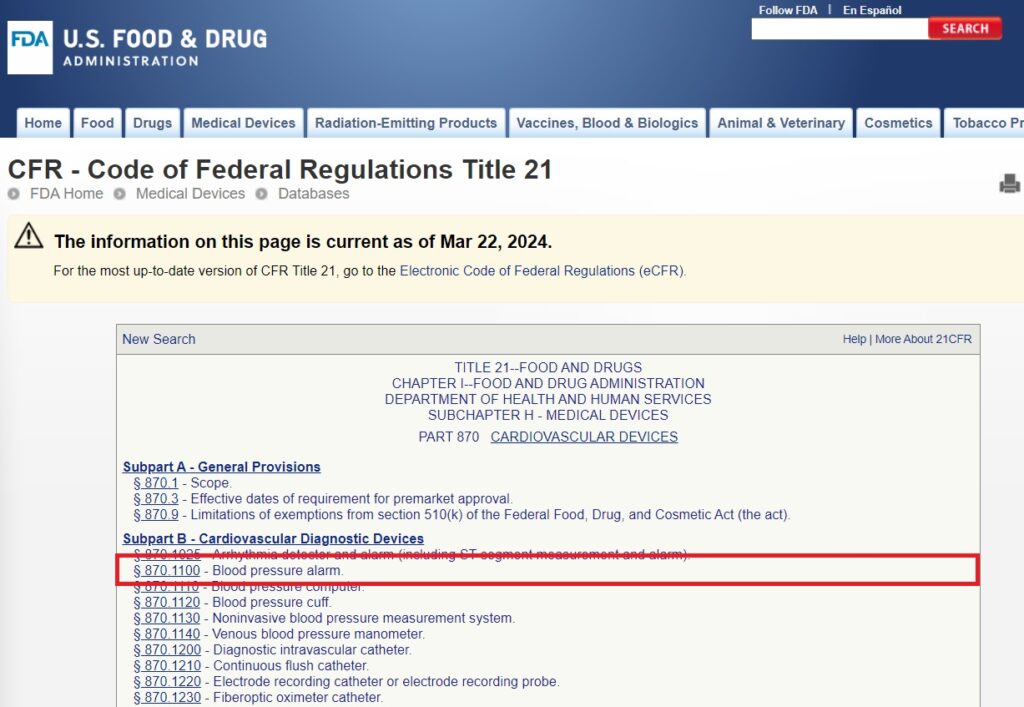
5. Review the Classification Guidelines: Click on the device code to access the detailed guidelines. Pay close attention to section (b), which specifies the device classification (Class I, II, or III).
6. Determine Risk Level and Controls: Based on the classification, understand the risk level associated with your device and the regulatory controls that apply:
- Class I: Low risk, general controls
- Class II: Moderate risk, general and special controls
- Class III: High risk, general controls, and premarket approval
7. Consider Novel Devices: If your device doesn’t match any of the approximately 1,700 devices classified by the FDA, it may be considered an innovative product without a substantial equivalent. In such cases, your device is likely to be classified as Class III by default.
8. Seek Expert Advice: If you’re unsure about the classification or your device seems to fall between categories, consider consulting with regulatory experts or contacting the FDA directly for guidance.
9. Prepare for Next Steps: Once you’ve determined the classification, begin preparing for the appropriate regulatory pathway:
- Class I: Most are exempt from premarket notification
- Class II: Usually requires 510(k) submission
- Class III: Typically requires Premarket Approval (PMA)
10. Stay Informed: Keep in mind that classifications can change. Stay updated on FDA regulations and guidance documents related to your device type.
Remember, accurate classification is essential as it determines the regulatory requirements your device must meet. Misclassification can lead to delays, additional costs, or regulatory issues down the line. When in doubt, it’s always better to err on the side of caution and seek professional guidance to ensure compliance with FDA regulations.
For manufacturers navigating the FDA medical device classes, partnering with an experienced medical device manufacturer is crucial for success. Arterex Medical, a leading manufacturer in the medical device and life-sciences industries, offers comprehensive solutions across all device classifications.
From initial Medical Device Design and Engineering through Medical Device Prototyping and 3D Printing, our expertise spans the full development cycle.
Our capabilities in Medical Device Contract Manufacturing, Medical Device Injection Molding, and Electromechanical Medical Device Design and Manufacturing enable them to produce devices across all FDA classes. For specialized needs, they excel in Single-Use Medical Device Manufacturing while ensuring regulatory compliance.
Their Medical Device Product Transfers and Medical Device Supply Chain Management services help manufacturers maintain consistent quality and regulatory compliance throughout the product life cycle. Whether developing a Class I, II, or III device, having a knowledgeable manufacturing partner can significantly streamline the path to market while ensuring adherence to FDA requirements.
- What are FDA Medical Device Classes?
- What Are Examples of Medical Devices?
- What Are The Different Types of Medical Devices?
- What is an FDA Class 1 Medical Device?
- What is an FDA Class 2 Medical Device?
- What is an FDA Class 3 Medical Device?
- What Is the Difference Between Class 1, Class 2, and Class 3 Medical Devices?
- How To Classify a Medical Device?