Medical Device Design Controls: Definition, Requirements, Process, Flow Chart, Phases, and Examples
Written by
Arterex Medical
Published on
June 26, 2025
Read time
15 minute read
Design controls are a systematic set of requirements and procedures that govern the design and development process for medical devices. They form a critical component of the quality management system required by regulatory bodies worldwide. These controls ensure that medical devices are safe, effective, and meet the needs of users and patients before they reach the market.

In essence, design controls provide a framework that transforms innovative ideas into manufactured medical devices through a documented, traceable, and verifiable process.
What Are Medical Device Design Controls?

Medical device design controls are a set of formalized procedures and documentation practices used during the product development lifecycle to ensure that medical devices are safe, effective, and meet user needs. These controls are mandated by regulatory bodies such as the FDA (21 CFR 820.30) and ISO 13485, particularly for Class II and Class III devices.
Design controls help manufacturers:
- Define product requirements clearly
- Minimize design-related risks
- Ensure compliance with regulatory standards
- Produce consistent and high-quality devices
The design control process includes several key phases:
- Design planning
- Design inputs and outputs
- Design reviews
- Verification and validation
Without proper design controls, companies risk product recalls, regulatory penalties, and patient harm.
What is the Importance of Traceability in Medical Device Design Control?
Traceability in medical device design refers to the ability to link each stage of product development — from user needs to design inputs, outputs, testing, and final validation. It ensures that every requirement has been addressed and verified through proper documentation and testing.
Why Traceability Matters?
- Regulatory Compliance: Traceability is essential for passing audits and achieving FDA or CE mark approvals.
- Risk Management: Helps identify and resolve issues early in the design process.
- Change Control: Makes it easier to assess the impact of design changes.
- Accountability: Maintains clear records of who did what and when.
- Efficiency: Speeds up reviews, approvals, and troubleshooting.
User Need | Design Input | Design Output | Verification Method | Validation |
Easy to use interface | UI must respond within 1 sec | Frontend interface code | UI response time test | Usability testing |
What Are Medical Device Design Control Examples?
Here are practical examples of how design controls are applied during medical device development:
1. Design Input
- Device Type: Blood Glucose Monitor
- Input: Must provide accurate readings within ±10% of laboratory values
- Source: ISO 15197 standard + user needs
- Why It Matters: Guide the accuracy standards the design must meet
2. Design Output
- Device Type: Infusion Pump
- Output: Electrical schematic of control board and software logic
- Why It Matters: The physical blueprint that matches the input requirements
3. Design Verification
- Device Type: Wearable ECG Monitor
- Test: Bench test confirms signal accuracy against predefined parameters
- Purpose: Verifies that the design output meets the input requirements
4. Design Validation
- Device Type: Digital Thermometer
- Test: Real-world testing with users in clinical environments
- Purpose: Ensures the device meets user needs and functions under expected use
5. Design Change
- Change: Switch in material used for housing a device
- Action: Assess impact on biocompatibility, repeat verification and validation
- Documentation: Update DHF and traceability matrix
Why Are Medical Device Design Controls Important?
Medical device design controls are crucial because they ensure that every product developed is safe, effective, and compliant with regulatory standards. They serve as a structured roadmap for taking a product from concept to commercialization while minimizing risks and errors.
Here’s why design controls are so important:
1. Ensure Patient Safety and Product Quality
Design controls help identify and address potential design flaws before the device reaches the market. By validating that the device meets user needs and performs as intended, manufacturers can prevent failures that could harm patients or users.
2. Regulatory Compliance
Agencies like the FDA (21 CFR 820.30) and organizations like ISO (ISO 13485) require design controls for most medical devices. Failing to implement proper design controls can result in product recalls, warning letters, import bans, or denial of regulatory approval.
3. Facilitate Traceability and Documentation
Design controls enforce disciplined documentation practices throughout the development lifecycle. This enables complete traceability — linking design inputs to outputs, verification, and validation. Such traceability is essential for audits and regulatory submissions.
4. Reduce Costly Redesigns and Delays
By identifying risks and design errors early, design controls help avoid expensive and time-consuming rework. Proper reviews, verifications, and validations ensure the device is built right the first time, reducing development time and costs.
5. Streamline Market Approval and Launch
A well-documented design control process increases the likelihood of a successful FDA 510(k) submission or CE marking. It shows regulators that your product development process is robust, controlled, and compliant with international standards.
6. Support Risk Management and Continuous Improvement
Design controls work hand-in-hand with risk management (e.g., ISO 14971), helping teams identify, assess, and mitigate risks systematically. This leads to safer, more reliable products and supports continuous improvement in future designs.
What Are the Medical Device Design Control Requirements?
Medical device design control requirements are regulatory guidelines that ensure medical devices are developed to meet user needs, intended use, and regulatory compliance. These structured processes are crucial for guaranteeing safety, effectiveness, and quality throughout the product lifecycle. The primary standards that define design control requirements are:
- FDA 21 CFR 820.30 (U.S. regulatory framework)
- Regulation (EU) 2017/745 (EU MDR) (European regulatory framework)
- ISO 13485:2016 / EN ISO 13485:2016 (International quality management system standard)
FDA 21 CFR Part 820 Design Control Requirements
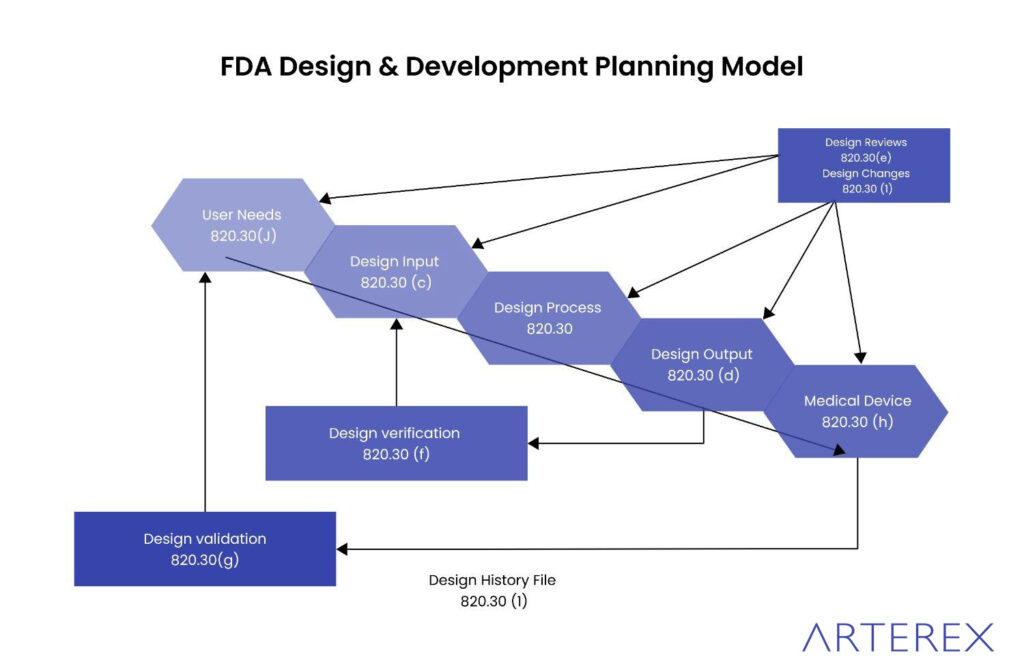
The U.S. FDA outlines its design control requirements in 21 CFR Part 820.30, which is part of the broader Quality System Regulation (QSR). These requirements apply to Class II and Class III medical devices marketed in the United States and ensure devices meet both user and regulatory expectations.
Key Elements of 21 CFR 820.30:
- Design and Development Planning
Manufacturers must create and maintain plans that define responsibilities, tasks, and timelines for design activities. - Design Input
Establish and document design requirements that consider the device’s intended use and user needs. Inputs must be reviewed and approved. - Design Output
Outputs must be measurable, meet input requirements, and be documented with clear acceptance criteria. - Design Review
Conduct formal reviews at defined stages of the project involving cross-functional stakeholders. All outcomes must be documented. - Design Verification
Confirm that design outputs meet design inputs. Results must include methods, participants, and dates. - Design Validation
Validate that the device meets user needs and intended use under real or simulated use conditions. Software and risk analysis are also included. - Design Transfer
Ensure the design is accurately translated into production specifications. - Design Changes
Implement a controlled process for documenting, evaluating, validating, and approving design changes. - Design History File (DHF) Maintain a DHF for each device, documenting that the design was developed in accordance with the approved plan and 21 CFR 820.30.
Regulation (EU) 2017/745 (EU MDR)
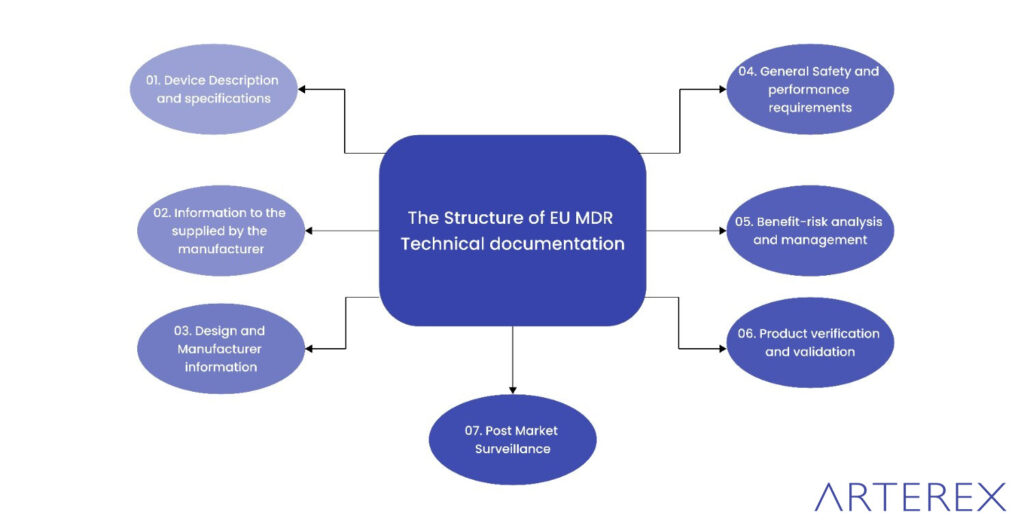
The EU Medical Device Regulation (EU MDR) 2017/745 provides a comprehensive legal framework for ensuring the safety and performance of medical devices marketed in the European Union. While the regulation itself does not break down design controls into individual steps like the FDA’s 21 CFR 820.30, it requires manufacturers to establish and maintain a robust Quality Management System (QMS).
Under Article 10 of the EU MDR, manufacturers are obligated to implement a QMS that governs all aspects of product realization. This includes—but is not limited to—the following key areas relevant to design control:
1. Design and Development
Manufacturers must implement procedures to control the design and development of devices. This involves:
- Translating user needs and regulatory requirements into design inputs,
- Planning design stages and verification/validation activities,
- Documenting changes and conducting formal design reviews.
While EU MDR doesn’t detail these steps, compliance is achieved by following EN ISO 13485:2016 Section 7.3, which defines structured requirements for managing design throughout its lifecycle.
2. Production and Service Provision
This step ensures that once the design is finalized, it is effectively transferred into manufacturing with consistent quality. It involves:
- Validating production processes,
- Establishing work instructions and acceptance criteria,
- Ensuring traceability and documentation throughout production.
This aligns with Section 7.5 of ISO 13485:2016.
3. Risk Management
Manufacturers are required to establish a continuous risk management process in accordance with ISO 14971. Within the design phase, this includes:
- Identifying potential hazards related to the device and its use,
- Estimating and evaluating associated risks,
- Implementing risk controls and verifying their effectiveness,
- Documenting the entire risk management file.
Risk considerations must be integrated into each stage of design and development.
4. Clinical Evaluation
A thorough clinical evaluation is mandatory to demonstrate the device’s safety and clinical performance. This involves:
- Reviewing relevant scientific literature,
- Analyzing clinical data from similar devices,
- Conducting clinical investigations if necessary,
- Ensuring that the design meets intended medical benefits with an acceptable risk-benefit ratio.
Clinical evaluation must be continually updated throughout the product’s lifecycle.
To meet these requirements effectively, manufacturers should align their internal QMS with EN ISO 13485:2016, particularly:
- Section 7.3: Design and Development
- Section 7.5: Production and Service Provision
- Section 4.1.2: Integration of Risk Management
- Annex Z of ISO 13485, which maps its clauses to EU MDR requirements
By doing so, organizations can ensure their design control processes meet EU MDR expectations—even if the regulation does not explicitly detail each step.
ISO 13485:2016 Design Control Requirements
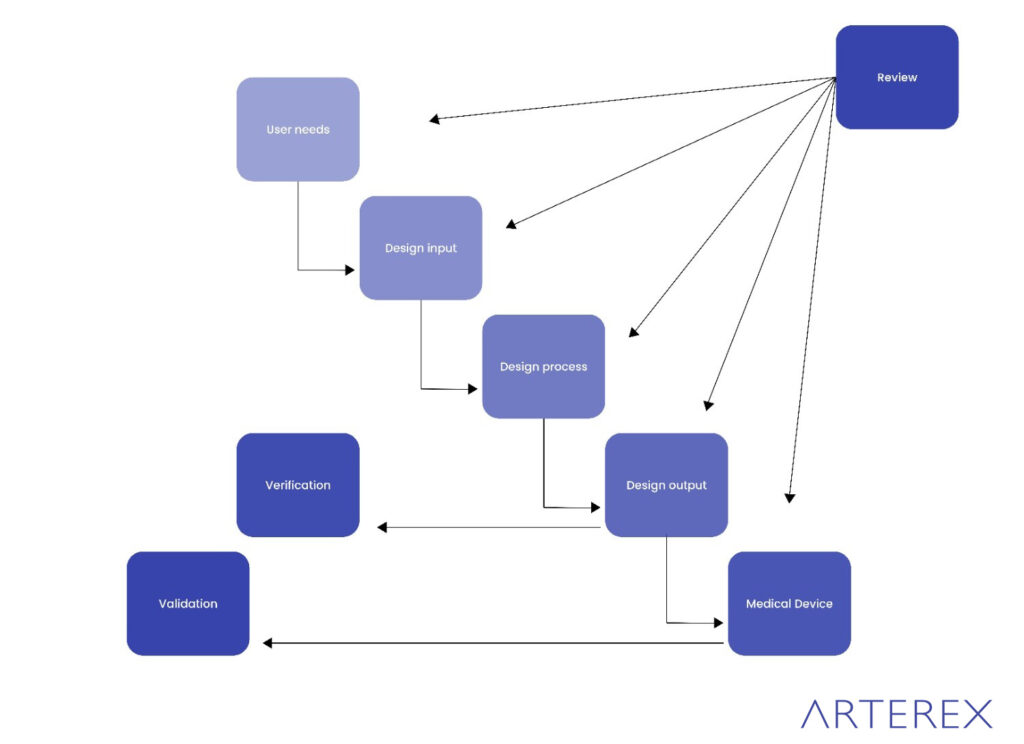
ISO 13485:2016 is the internationally recognized standard for medical device quality management systems. It ensures consistent compliance with customer and regulatory requirements across the product lifecycle—from development to post-market activities.
Section 7.3 of ISO 13485:2016 – Design and Development
- Planning of Product Realization (7.1)
Define objectives, allocate resources, establish validation steps, and ensure traceability. - Customer-Related Processes (7.2)
Translate user and regulatory requirements into actionable design and manufacturing specifications. - Design and Development (7.3)
Implement structured controls, including design reviews, verification, validation, and change management. - Purchasing (7.4)
Validate supplier compliance and component quality. - Production and Service Provision (7.5)
Use validated procedures to ensure consistent manufacturing and servicing standards. - Control of Monitoring and Measuring Equipment (7.6)
Maintain and calibrate tools to ensure measurement accuracy and traceability.
While ISO 13485 does not mandate a Design History File (DHF), it requires robust documentation and traceability equivalent to FDA requirements.
Simplify Medical Device Design Controls with Expert Engineering
Turn complex design control requirements into a streamlined, audit-ready development process with Arterex Medical’s design and engineering expertise.
From design inputs and risk management to verification, validation, and design transfer, we support every phase of your product lifecycle.
Ensure compliance with FDA 21 CFR 820.30, ISO 13485, and EU MDR while accelerating time-to-market and reducing costly redesigns.
Explore Design & Engineering Services →What Is the Difference Between ISO 13485 and 21 CFR 820 Design Control Requirements?
The Difference Between ISO 13485 and 21 CFR 820 Design Control Requirements are explained in the table below:
Aspect | FDA 21 CFR 820 | ISO 13485:2016 |
Regulatory Scope | Legally enforced by the FDA in the U.S. | International standard used globally, including the EU |
Terminologies | Specifies DHF, design input/output, verification, validation, etc. | Covers the same concepts without rigid terminology like “DHF” |
Risk Management | Encouraged through industry best practices | Explicitly integrated with ISO 14971 |
Country of Application | Mandatory in the U.S. | Used for global and EU compliance (e.g., CE marking) |
Documentation | Requires DHF to trace full design history | Requires equivalent documentation but no DHF term used |
The table below provides a side-by-side comparison of the design control clauses outlined in FDA 21 CFR 820.30 and those found in Section 7.3 of ISO 13485:2016.
FDA 21 CFR 820.30 – Design Control | ISO 13485:2016 Section 7.3 – Design and Development |
(a) General | 7.3.1 General |
(b) Design and development planning | 7.3.2 Design and development planning |
(c) Design input | 7.3.3 Design and development inputs |
(d) Design output | 7.3.4 Design and development outputs |
(e) Design review | 7.3.5 Design and development review |
(f) Design verification | 7.3.6 Design and development verification |
(g) Design validation | 7.3.7 Design and development validation |
(h) Design transfer | 7.3.8 Design and development transfer |
(i) Design changes | 7.3.9 Design and development changes |
(j) Design history file | 7.3.10 Design and development files |
In 2024, the FDA introduced a final rule to harmonize 21 CFR Part 820 with ISO 13485, renaming it the Quality Management System Regulation (QMSR). This effort aligns terminology and expectations, minimizing conflicts between U.S. and international regulations.
- The QMSR will take effect in 2026.
- Until then, manufacturers must comply with the current 21 CFR 820 regulations.
- Harmonization will ease dual compliance for manufacturers operating in both U.S. and global markets.
- The QMSR will take effect in 2026.
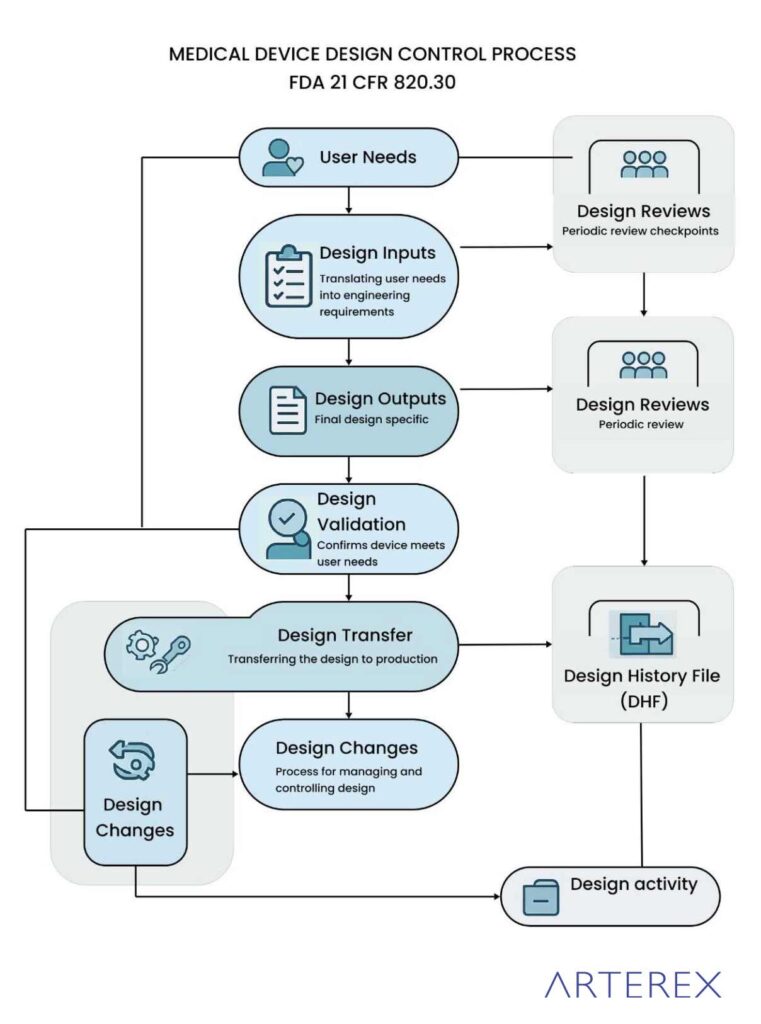
What is the Process of Medical Device Design Controls?
The medical device design control process is a structured framework that ensures a device is designed to meet user needs, intended use, and applicable regulatory requirements. This process is a critical part of the product development lifecycle and is required by regulatory bodies like the FDA (21 CFR 820.30), ISO 13485, and the EU MDR (Regulation (EU) 2017/745).
The design control process typically includes the following key phases:
1. Design and Development Planning
This initial phase involves creating a comprehensive plan that outlines all design activities, assigns responsibilities, and defines interdepartmental communication. The plan must be reviewed and updated as the project progresses.
2. Design Inputs
Design inputs are the foundation of the product’s development. These include user needs, intended use, regulatory requirements, performance specifications, and risk management considerations. Inputs must be documented, reviewed, and approved before proceeding.
3. Design Outputs
Design outputs are the tangible results of design efforts. They may include drawings, specifications, manufacturing instructions, and quality assurance procedures. Outputs must align with design inputs and include acceptance criteria.
4. Design Review
Formal, documented design reviews are conducted at various stages to evaluate progress, identify issues, and ensure that design requirements are being met. These reviews must involve representatives from multiple functional areas and must be recorded.
5. Design Verification
Verification ensures that the design outputs meet the specified design inputs. This phase involves testing, inspection, and analysis using defined methods. All verification activities and results must be documented.
6. Design Validation
Validation confirms that the final product meets user needs and intended uses under actual or simulated conditions. It includes clinical evaluation, usability testing, and software validation (if applicable). This step must be completed before product release.
7. Design Transfer
This phase ensures that the device design is accurately and efficiently translated into production specifications. It involves coordination between design and manufacturing teams to ensure reproducibility and product quality.
8. Design Changes
Any changes to the design must be systematically controlled. This includes evaluating the impact, documenting the change, verifying and validating as necessary, and obtaining approvals before implementation.
9. Design History File (DHF)
The DHF is a compilation of all records generated throughout the design process. It demonstrates that the device was developed according to the approved design plan and regulatory requirements.
Medical Device Design Control Process flow chart
Integrating Risk Management in Design Control Process
Integrating risk management into the design control process is critical to ensuring medical device safety and regulatory compliance. Risk management is not a one-time task but a continuous process that spans the entire product lifecycle. Standards like ISO 14971 guide manufacturers on how to identify, assess, control, and monitor risks associated with medical devices.
Ties Between ISO 14971 and Design Controls
ISO 14971 is the internationally recognized standard for medical device risk management. It complements design control requirements outlined in FDA 21 CFR 820.30 and ISO 13485:2016, making it an essential part of the design and development framework.
Design controls require verification and validation of design outputs, while ISO 14971 ensures risks are systematically evaluated and controlled. Both standards emphasize documenting decision-making processes and outcomes related to product safety.
Risk Analysis During Design Phases
Risk analysis should be embedded into every phase of the design process—from initial concept to design validation. During these phases, manufacturers must:
- Identify potential hazards associated with the device’s intended use
- Estimate the probability and severity of harm
- Assess risks individually and in aggregate
- Decide whether risk reduction is necessary and how it can be achieved
Common risk analysis tools include Failure Modes and Effects Analysis (FMEA), Fault Tree Analysis (FTA), and Hazard Analysis and Critical Control Points (HACCP).
Documenting Risk Mitigation Strategies
All identified risks and corresponding mitigation strategies must be clearly documented. This includes:
- Describing the identified risk
- Specifying the control measures (e.g., design changes, protective measures, user training)
- Verifying the effectiveness of each mitigation
- Updating the risk management file as design changes occur
This documentation becomes part of the Design History File (DHF) and the Risk Management File, ensuring traceability and transparency.
What Are The Best Practices for Effective Implementation of Design Controls?
Effective implementation of design controls and risk management requires a proactive, structured approach. Here are key best practices:
Start Early in Product Development
Begin design controls and risk assessments during the concept or feasibility stage. Early integration helps identify issues before significant resources are invested and reduces the need for costly redesigns.
Use Design Control Templates and Tools
Utilize standardized templates for design inputs, outputs, risk analysis, verification, and validation. Tools like design control matrices and traceability matrices help map design elements to risk and compliance requirements, simplifying audits and reviews.
Involve QA/RA Teams from the Beginning
Quality Assurance (QA) and Regulatory Affairs (RA) teams play a critical role in ensuring that design and risk activities meet regulatory standards. Their early involvement promotes compliance, reduces delays, and strengthens overall product quality.
Maintain a Living Design History File (DHF)
The DHF should be continuously updated throughout the development process. Treat it as a “living” document that captures all changes, reviews, test results, and risk assessments. Keeping it current facilitates smoother regulatory submissions and demonstrates design control compliance during audits.
- Medical Device Design Controls: Definition, Requirements, Process, and Phases
- What Are Medical Device Design Controls?
- What is the Importance of Traceability in Medical Device Design Control?
- What Are Medical Device Design Control Examples?
- Why Are Medical Device Design Controls Important?
- What Are the Medical Device Design Control Requirements?
- FDA 21 CFR Part 820 Design Control Requirements
- Regulation (EU) 2017/745 (EU MDR)
- ISO 13485:2016 Design Control Requirements
- What Is the Difference Between ISO 13485 and 21 CFR 820 Design Control Requirements?
- What is the Process of Medical Device Design Controls?
- Medical Device Design Control Process flow chart
- Integrating Risk Management in Design Control Process
- What Are The Best Practices for Effective Implementation of Design Controls?